(This is a preview of the hypothetical disease model being described in the upcoming paper.)
(Click here to explore the experimental intervention protocol based on this work.)
These figures are a draft and may contain errors. An interactive version for some of these diagrams is being actively developed and a beta version can be found here. It is currently not working on mobile browsers; however, you can also right-click on the images below and open them in a new tab / save them for high resolution and/or easier navigation.
Questions / discussions - please join our Discord server - https://discord.gg/CAMXV78 (the forum previously hosted here was relocated to Discord in 2021).
(New, experimental): You can also navigate the disease model and protocol using an AI assistant - Google's NotebookLM. A setup guide can be found here.
The initial presentation of the model from February 2023 and various others can be found on the Videos page.
The most recent presentation, as of July 2025 can be found below:
Figure 1 (click image to download PDF) or use the Metabolic Pathway Overlay tool to view this diagram with optional data overlay:

Figure 2 (legacy material - being retired/superseded by Figure 3 and will be replaced soon):

Figure 3: (Interactive explorer - here)

Figure 4 (click image to download PDF and interactive explorer - here)
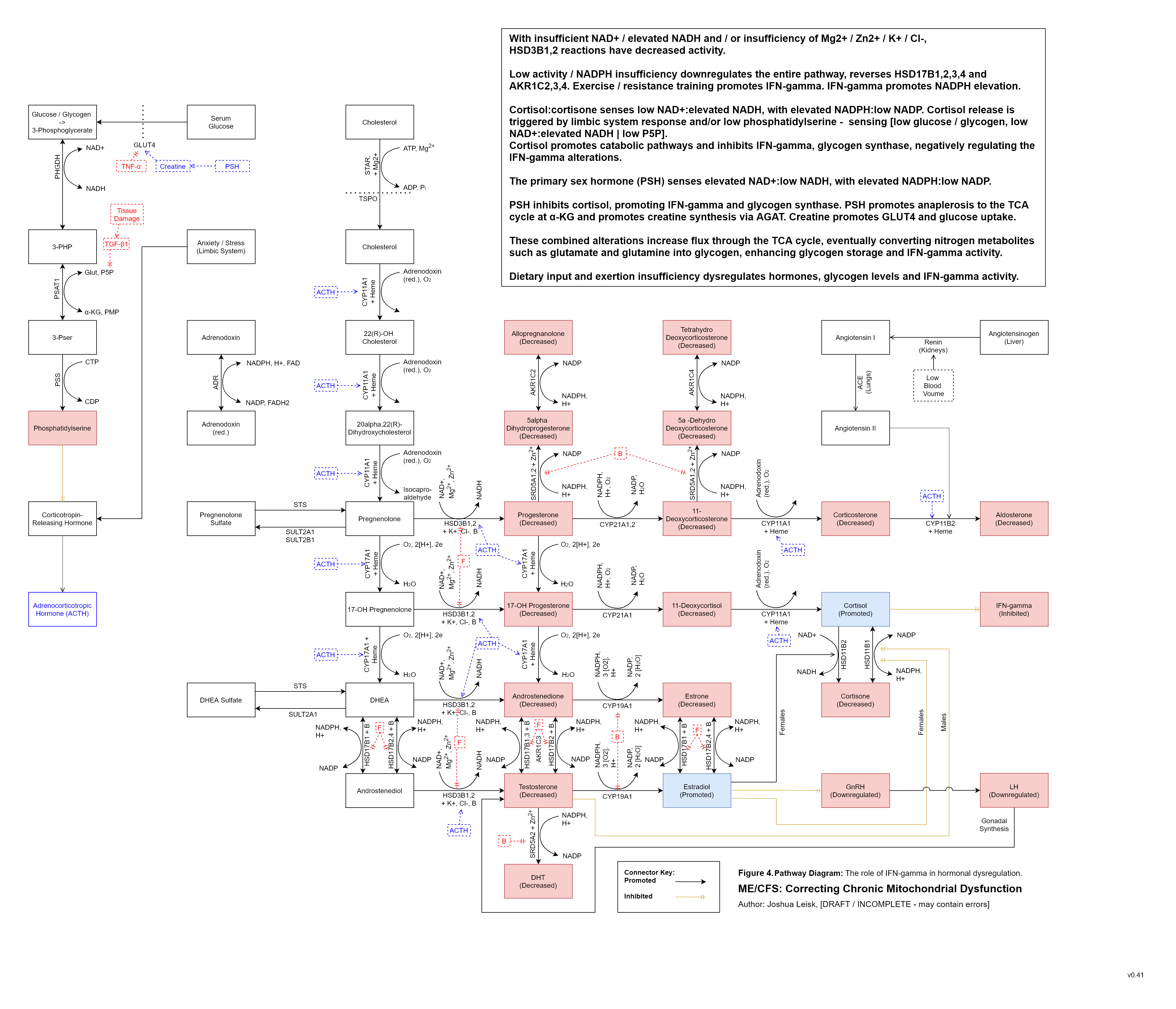
Figure 5 (Interactive explorer - here):
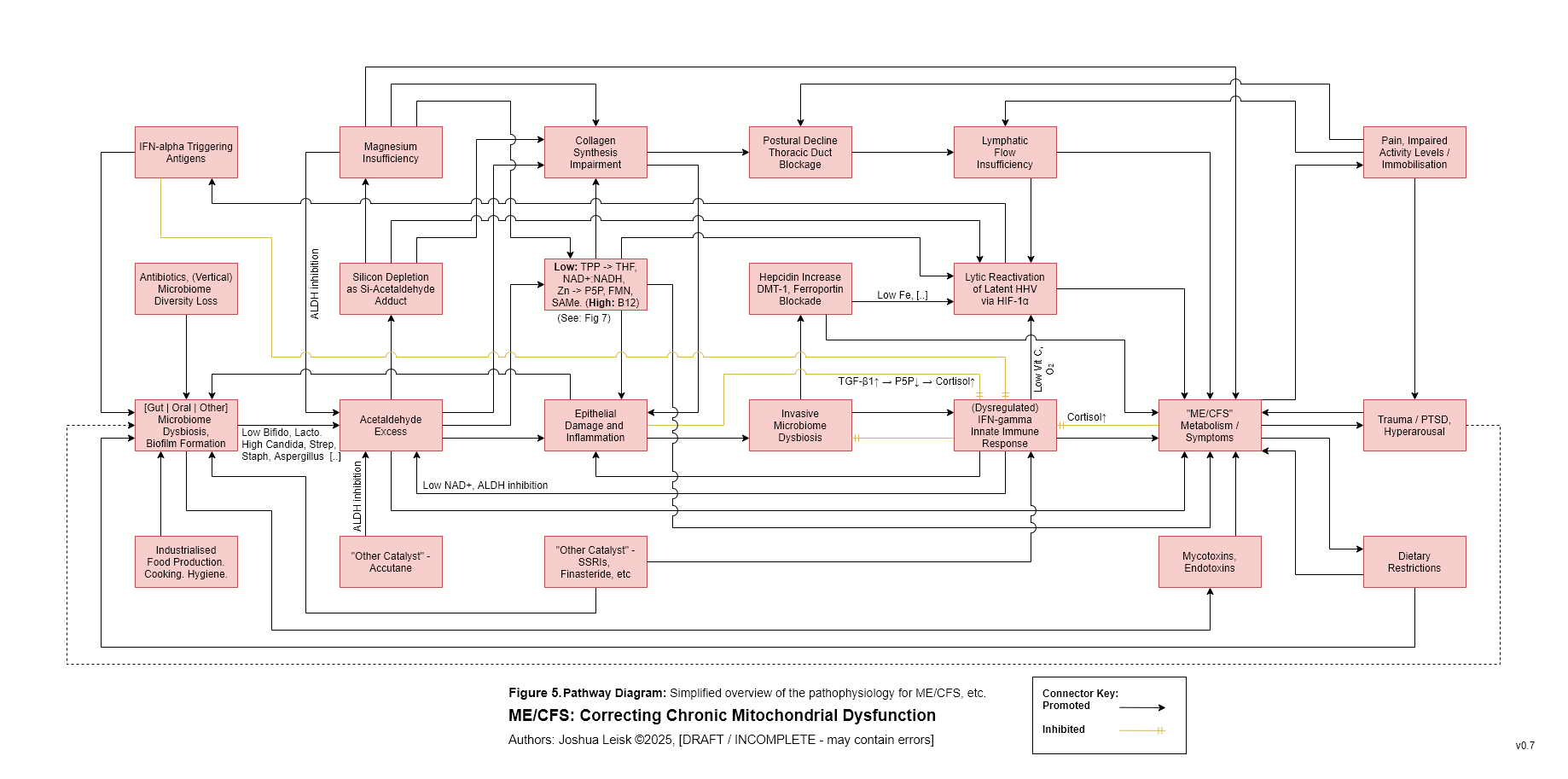
Figure 6 (click image to download PDF):
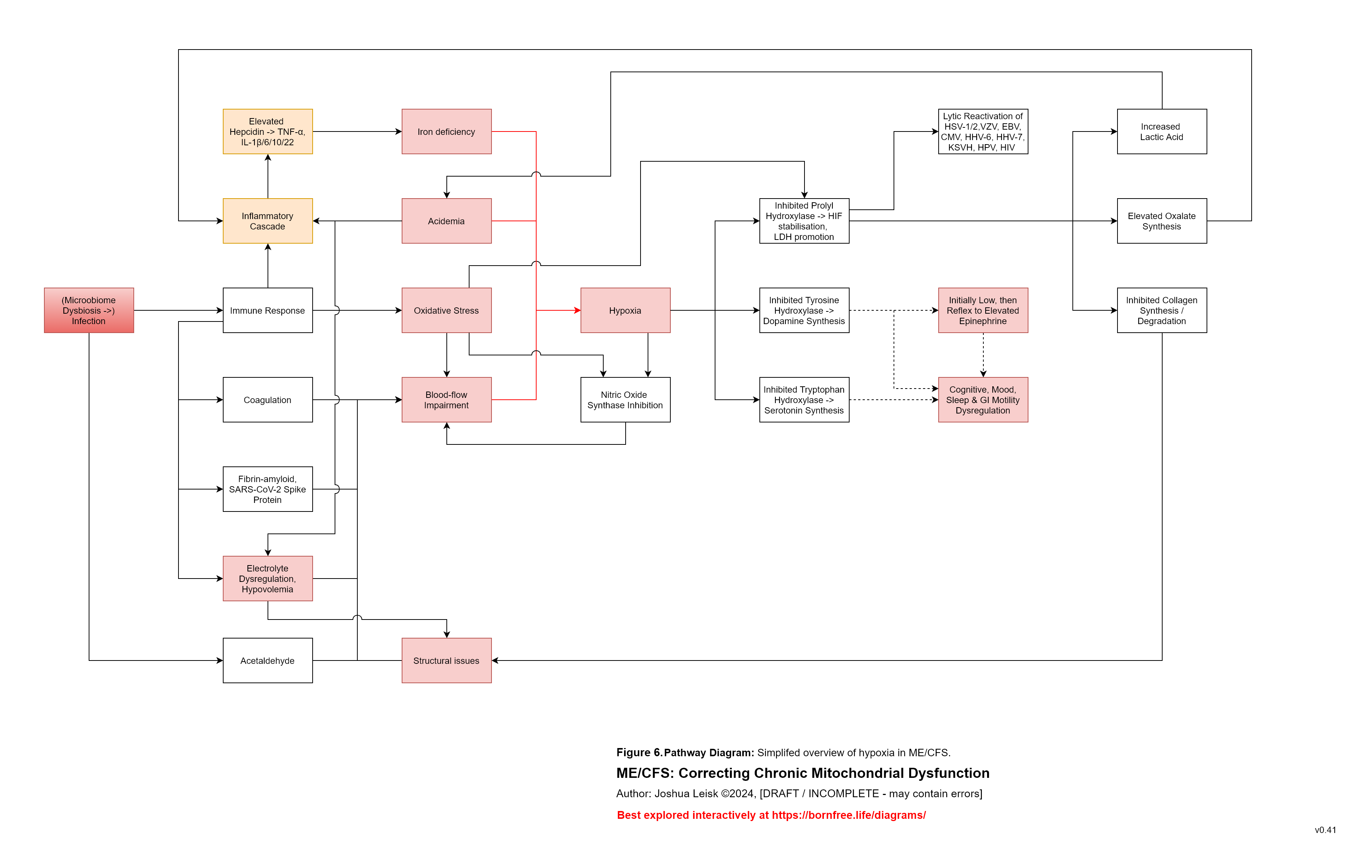
Figure 7 (click image to download PDF):

Figure 8 (click image to download PDF):

Figure 9 (click to download PDF):

Figure 10 (click image to download PDF):

Figure 11 (click image to download PDF):

Figure 12 (click image to download PDF):

Figure 13 (click image to download PDF):

Figure 14 (click image to download PDF):

Figure 15 (click image to download PDF):

Figure 16 (click image to download PDF):

Figure 17 (click image to download PDF):
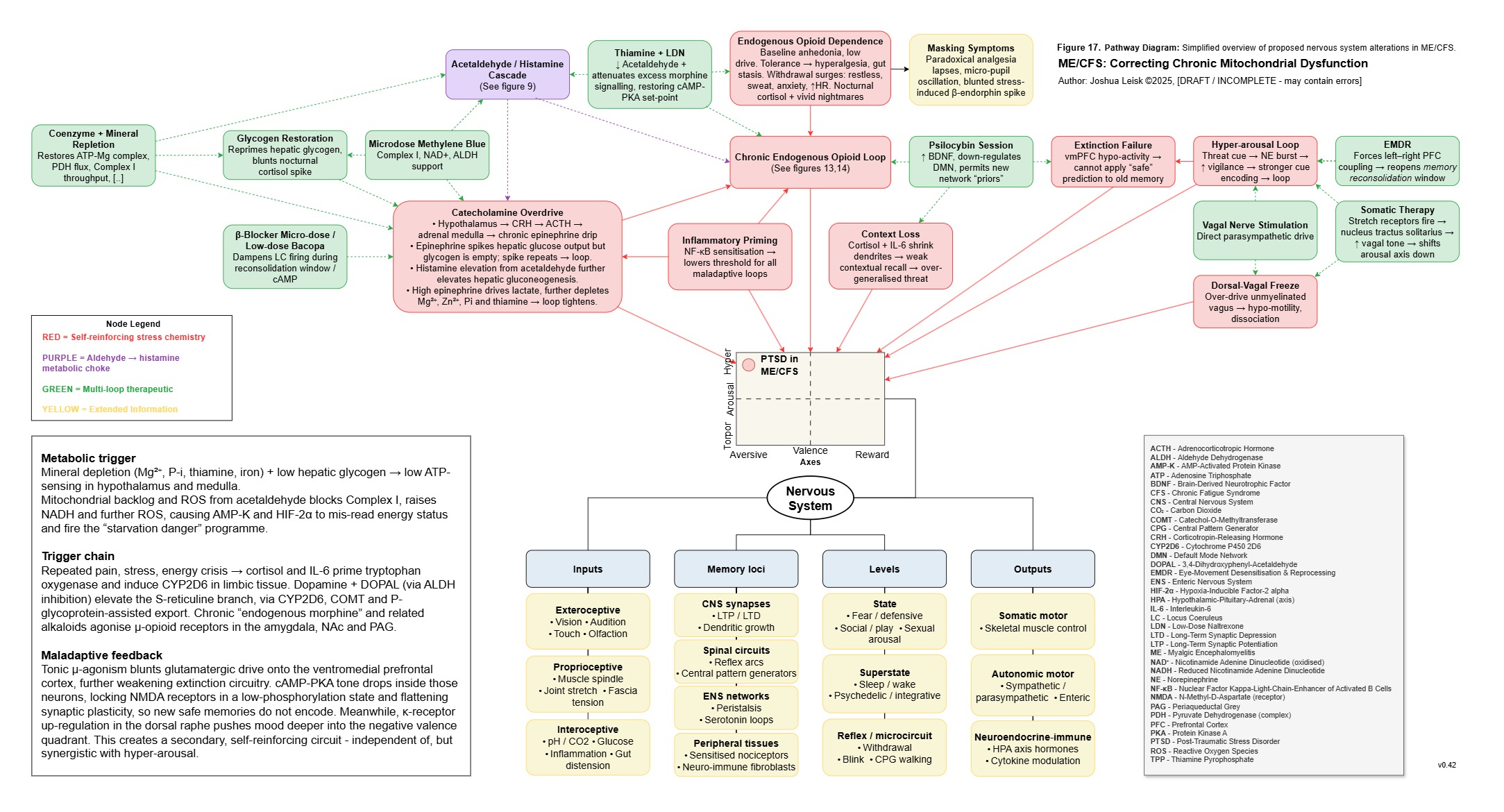
Figure 18 (click image to download PDF):

Figure 19 (click image to download PDF):

1. Biofilms are a protective 3D extracellular matrix surrounding microorganism and are a normal feature of the microbial lifecycle and our microbiomes - which can largely be found in any/all mucosal tissues.
A blind spot in the human immune system exists around the contents of these biofilms, allowing for “pathogenic” species to create hidden reservoirs.
Pathogenic species of particular interest to this disease model include acetaldehyde producing species, such as H. pylori, S. aureus, Streptococcus spp., Klebsiella spp., E. coli, Candida spp., Aspergillus spp. and others.
The contents of biofilms are not inspected and therefore an immune checkpoint is only provided during the planktonic and yeast stages of the microbial lifecycles, preventing biofilm surface area expansion.
These biofilms are also degraded by helpful microorganisms (various Lactobacillus spp., Bifidobacterium spp., Bacillus spp. and other species), stomach acid, dietary intakes of biofilm degrading compounds and lifestyle choices.
2. Through environmental exposure, small amounts of pathogenic biofilms will form in very early childhood. Moreso, if the microbiome has been compromised by a lack of protective species, such as Bifidobacterium spp. and Lactobacillus spp.
These protective species are abundant in human breast milk and can make up 80% of a neonate’s gut microbiome and 20% of a “healthy” adult human’s gut microbiome. Conversely, low populations of bifidobacterium are routinely found in chronic disease.
These protective microorganisms metabolise acetaldehyde into acetate, degrade biofilms and inhibit pathogenic species.
Breast milk contains calcium and phosphorus to help buffer stomach acid pH and allow survival of these protective species through the stomach and small intestine. Breast milk contains selective prebiotics such as lactose and human milk oligosaccharides, supporting colonisation of these species. Milk also contains colostrum, lactoferrin, xanthine oxidase and IgG, supporting mucosal immunity.
Antibiotics have broadly deleterious effects on microbiome diversity and abundance.
3. An ongoing slippery slope of microbiome dysbiosis is created by events which allow the surface area expansion of pathogenic biofilms. These events may include introduction of a catalyst or antigen which distracts/dysregulates immune activity or decreases the diversity and counts of protective microorganisms;
eg. Immune system dysregulation caused by malnutrition, low NK cell counts, medications that alter Interferon signalling bias / cascade regulation, antibiotics, biotoxins, IFN-alpha promoting antigen dominance (eg. SARS-CoV-2 spike protein / infection, influenza, reactivated herpesviruses, isocitrate lyase expressing microorganisms, lipopolysaccharides, medical interventions which provide antigens for immune imprinting), and chronic stress (as elevated cortisol, dysregulation of cytokines) can each provide a window of opportunity for unimpeded biofilm growth, increased endo/mycotoxin production and net acetaldehyde increase.
Acetaldehyde is well-known in chronic alcoholism for causing T-cell exhaustion, inhibiting glycolysis, decreasing NAD+/elevating NADH, inhibiting methylation, inhibiting collagen synthesis, dysregulating thiamine pyrophosphate metabolism and having a higher affinity for various aldehyde dehydrogenase (ALDH) isoenzymes required for eg. neurotransmitter degradation, histamine degradation and many other pathways.
Gliotoxin, other mycotoxins and endotoxins are also relevant to this cascade.
4. These and other factors can lead to degradation of the epithelium, chronic low-level infection / bacteraemia / fungaemia and chronic innate immune response + mast cell activation, as the pathogenic reservoirs are hidden from the immune system.
An inflammatory cascade involving TNF-alpha, IL-1beta /6/10/22 promotes hepcidin. Hepcidin is a peptide-hormone which inhibits divalent metal transporter-1 (DMT-1) and ferroportin. These 2 transporters are well-known for regulating systemic iron homeostasis, however the literature shows they are also responsible for 8 other metals required in energy metabolism, neurotransmission, collagen synthesis and other relevant pathways.
Enterocytes in the gut express both DMT-1 and ferroportin. Cells in the brain, liver and kidneys only express ferroportin. This means that while the absorption of dietary minerals is blockaded in the gut, circulating minerals are sequestered in brain, liver and kidney tissues until the inflammatory cascade subsides.This mineral transport blockade appears to benefit the host in an acute infection by limiting available minerals for pathogen proliferation, however in chronic infection leads to induced malnutrition / mineral depletion.
5. The IFN-gamma innate immune response alters transcription factors and NAD+ metabolism in specific ways which favour immune activity against microbial and other pathogens.
An elevation of IFN-gamma signalling can result in reactive oxygen species (ROS) generation by at least 3 enzymes - xanthine oxidase (XO), NADPH oxidase (NOX) and nitric oxide synthase (NOS). NOS is also a reactive nitrogen species generator when tetrahydrobiopterin (BH4) is low.
IFN-gamma inhibits mitochondrial complex I / NADH dehydrogenase, inhibiting the NAD+ redox and allowing the NADH pool to increase.
Increased NADH pool status from TCA cycle flux created by exertion / metabolic activity promotes NADP transhydrogenase activity in converting NADP into NADPH, returning one NAD+.
NAD+ biosynthesis is simultaneously upregulated at IDO1/TDO, enhancing catabolism of tryptophan through the kynurenine pathway, however this is limited by a number of factors, some highlights for which include (endogenous and microbial synthesis of) tryptophan, P5P pool status and oxidative stress.
Acetaldehyde metabolism by ALDH decreases NAD+ and promotes NADH.NAD+ and NADPH are used by a number of pathways to benefit host response to infection and tissue repair / adaptions:
For IFN-gamma activity’s ROS generation and oxidation of pathogens - XO requires elevated NAD+:NADH. NOX requires elevated NADPH: low NADP. NOS requires elevated NADPH:low NADP.
For methylation - methylenetetrahydrofolate reductase (MTHFR) and dihydrofolate reductase (DHFR) are promoted in one direction by elevated NADPH: low NADP, increasing 5-MTHF and BH4 recycling, respectively.
Hormone and cholesterol biosynthesis are promoted by elevated NADPH:low NADP and elevated NAD+: low NADH. Cortisol is decreased.
Conversely, cortisol inhibits IFN-gamma signalling / activity and is promoted by elevated NADH: low NAD+, low pyridoxal 5-phosphate (P5P) and low glycolytic flux. Cortisol appears to be the primary regulator for IFN-gamma activity, promoting catabolic energy pathways and allowing the various pool status to reset / replenish. The model predicts inversion of the diurnal cortisol release profile where cofactor pool status is not well-supported.
6. Excessive or dysregulated IFN-gamma activity depletes NAD+ and causes oxidative stress, relative to exertion and/or immune activity.
The model describes oxidative stress as a key factor in post-exertional malaise (PEM).
Various mineral and electrolyte deficiencies, caused by dietary insufficiency, renal dysfunction and/or the described mineral transporter blockade impairs metabolism of ROS.
During upregulated IFN-gamma activity, metalloenzymes which metabolise reactive oxygen species are promoted to protect cells against the innate immune response.
These metalloenzymes include CuZnSOD (requiring copper and zinc), MnSOD (requiring manganese), catalase (requiring heme), glutathione peroxidase and reductase (requiring selenium + cofactors for riboflavin -> adeflavin (FAD) metabolism - Zn, Mo, Ca, I, Se, Heme and Mg.)
These mineral deficiencies create or exacerbate oxidative stress during IFN-gamma activity and leads to exhaustion of available antioxidants - ascorbic acid, tocopherols / tocotrienols, BH4, cobalamin II, melatonin, glutathione, alpha-lipoic acid and dietary sources of antioxidants.
7. Oxidative stress and insufficiency of these antioxidants has a significant impact on tissue damage, energy metabolism and neurotransmitter homeostasis.
Glycolysis is inhibited by oxidative stress at pyruvate kinase and other reactions.
Carnitine biosynthesis is inhibited by oxidative stress. Carnitine is required to shuttle longer chain fatty acids in fatty acid oxidation.
The TCA cycle is inhibited in multiple places by oxidative stress.
Tissue damage from oxidative stress and other influences leads to promotion of transforming growth factor beta-1 (TGF-b1) and inhibition of vitamin B6 -> P5P metabolism, with a broad impact on neurotransmitter metabolism and 140+ other pathways.
Specific influences affecting prolyl hydroxylase activity leads to Hypoxia Inducible Factors (HIF-1a) stabilisation / promoting, triggering anaerobic glycolysis (lactic acid metabolism) and reactivation of human herpesviruses, human papilloma virus, HIV, etc.
These influences include:
Impaired glycolysis / fatty acid oxidation, low Mg and other factors leading to low alpha-ketoglutarate.
Low riboflavin -> FAD and/or elevated IFN-alpha -> itaconate, affecting succinate dehydrogenase activity, leading to elevated succinate.
Insufficiency of prolyl hydroxylase cofactors - low Fe/Si | low Zn | low vitamin C (caused by oxidative stress from IFN-gamma activity) | hypoxia.
8. A significant conflict exists between IFN-gamma activity, leading to low NAD+, the mineral and electrolyte deficiencies and ALDH activity. ALDH requires NAD+ as a cofactor, along with Mo, Zn and Mg. This creates / exacerbates aldehyde toxicity and especially acetaldehyde toxicity.
Overall, this is only a brief outline for key-points in the disease model.
The sum of the alterations outlined here and numerous others detailed in the disease model leads to dysregulation of aldehyde metabolism, histamine metabolism, B6 degradation, mitochondrial and other energy metabolism, neurotransmitter metabolism, renal function, blood volume and temperature regulation, immune functions and more.
Variables inside the cascade, such as mineral and nutritional status, biofilm locations, microbiome dysbiosis and pathogenic species involved predict feature presentation and severity.
The disease model suggests this cascade could be loosely described as microbe-mediated scurvy and alcoholism, with extras. It has further implications for aging and longevity.