(This is a preview of the hypothetical disease model being described in the upcoming paper.)
(Click here to explore the experimental intervention protocol based on this work.)
The figures below the disease model brief are a working draft and may contain errors. You can click on these images below and download high-resolution PDFs, or explore many of them interactively - visualising patient data as a heat-style overlay, using the Metabolic Pathway Overlay tool.
Questions / discussions - please join our Discord server - https://discord.gg/CAMXV78 (the forum previously hosted here was relocated to Discord in 2021).
(New, experimental): You can also navigate the disease model and protocol using an AI assistant - Google's NotebookLM. A setup guide can be found here.
The initial presentation of the model from February 2023 and various others can be found on the Videos page.
A clinician training session from May 2026 can be found below:
A (highly simplified) description of pathophysiology key-points:
Mitochondrial dysfunction is associated with a wide range of chronic diseases, disorders and syndromes, such as Autism Spectrum disorders, ADHD, ME/CFS, Long COVID, Post Vaccination Syndrome, CIRS, Fibromyalgia, Lyme Disease and a growing number of age-related diseases. Coupling systemic mitochondrial dysfunction with localised infections, tissue-specific ionomic disturbances and incomplete healing may predict and explain a number of "named" chronic illnesses.This hypothetical model describes chronic mitochondrial dysfunction as a progressive failure of redox control, mineral handling, aldehyde detoxification, purine metabolism, histamine regulation, inflammatory signalling, connective tissue maintenance, microbiome stability and autonomic balance. It is not framed as a single-defect disease. The central claim is that persistence emerges when several normally compensable disturbances become mutually reinforcing, especially impaired NAD⁺/NADH and NADP⁺/NADPH redox balance, poor glutathione synthesis and recycling, dysregulated metal transport, intracellular glutamate elevation, extracellular ATP danger signalling, impaired glycogen metabolism, acetaldehyde burden, histamine excess and sympathetic-dominant physiology.
The model allows for vulnerability to begin before birth. In some cases, the system may already be biased by maternal metal burden, placental mineral transport, immune activation during pregnancy, altered maternal glucose handling, gestational stress physiology or inflammatory signalling. Gestational diabetes, in this context, is not interpreted only as excess glucose or conventional insulin resistance. It may also represent an early redox and storage problem, where glucose cannot be safely routed into glycogen synthesis, pentose phosphate pathway activity, NADPH regeneration, glutathione recycling and anabolic repair. If magnesium, phosphate, zinc, selenium, folate, choline, thiamine, riboflavin, carnitine or related cofactors are insufficient, hyperglycaemia may partly reflect impaired metabolic containment rather than merely carbohydrate excess. The foetal environment may therefore inherit not only nutrients and toxins, but also a redox pattern, mineral-transport context and inflammatory tone.
[see figure 14]
After birth, the model proposes an initial slow spiral before typically an abrupt disease onset event. Chronic stress, insufficient carbohydrate availability, low magnesium, phosphate, selenium, zinc, silica, folate or choline, infection, sleep disruption, neurodivergent stress load, trauma, connective tissue fragility and impaired collagen turnover may gradually narrow the system’s adaptive range. During this phase the organism may still function, but at increasing cost. NAD⁺ synthesis, NADP redox, glutathione production, methylation, transsulfuration, glycine availability, cysteine availability, glutamate clearance, zinc status, magnesium-dependent ATP chemistry and purine synthesis may all become progressively more fragile.
[see figure 14]
This slow spiral also creates conditions where toxic elements and essential minerals become progressively misallocated. Heavy metals and other disruptive elements become harder to neutralise or excrete if glutathione metabolism, metallothionein function, bile flow, renal handling and redox control are impaired. At the same time, essential electrolytes and trace minerals may become harder to retain in the correct compartments. The model therefore distinguishes between simple deficiency and pathological redistribution. A person may measure normal, elevated or decreased levels in serum while inversely having intracellular misallocation or organ-level sequestration and accumulation, depending on the mineral or metal, also creating challenges for standard diagnostics approaches.
[see figure 12]
A catalytic event may then push the system past threshold. This could be an infection, immune flare, toxin exposure, surgery, pregnancy, vaccination response, severe overexertion, fasting, heat stress, psychological trauma, sleep deprivation or another inflammatory insult. The proposed common pathway is elevation of inflammatory cytokines such as TNF-alpha, IL-1beta, IL-6, IL-10 and IL-22, with downstream hepcidin activation. Hepcidin then suppresses ferroportin and alters DMT1-related metal handling, changing the movement of iron and other divalent metals between blood, gut, liver, spleen, kidney, immune cells and brain. The key point is not merely that minerals become low, but that inflammatory signalling changes where they are allowed to go.
[see figure 12]
At this stage, the system can shift from compensation into metabolic collapse. Glycogenesis, glycolysis, pyruvate handling, TCA cycle flux, NAD⁺ regeneration, NADPH-dependent antioxidant defence and ATP-coupled mineral transport may all become constrained together. Mitochondrial capacity does not necessarily fail because the mitochondria are permanently destroyed, but because the surrounding regulatory environment no longer permits stable substrate flux, redox cycling, cofactor availability, antioxidant control or membrane transport. Major constraints in this context are now any excessive single or combination of specific toxic metals, inhibiting glutathione peroxidase (mercury, silver, gold), glutathione reductase (cadmium, lead) and glucose-6-phosphate dehydrogenase (aluminium), along with functional deficiencies of Mg-ATP (loop), glycine and cysteine and their upstream causes.
[see figure 14]
Resulting dysregulated intracellular glutamate elevation is released into the CSF and serum by the sympathetic signalling cascade, where elevated adrenaline triggers an elevation of the cAMP-PKA and glutamate efflux, causing pathological calcium influx and unsustainable cellular metabolic overdrive / excitotoxicity. Various other voltage-gated calcium channels, eg. TRPM3 are downregulated to compensate. Further adaptations, via eg. Substance P and TRPA1 are expected.
[see figure 18]
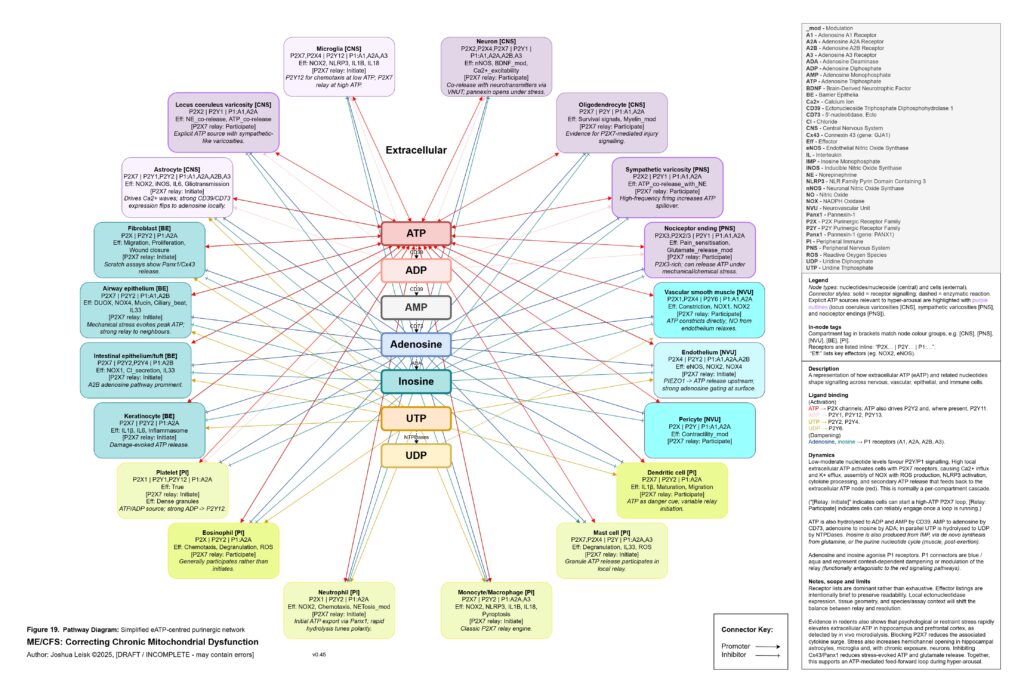
The extracellular purinergic system becomes central here. Under normal conditions, local ATP release is a useful signal. ATP, ADP, AMP, adenosine, inosine, UTP and UDP coordinate tissue repair, immune surveillance, vascular tone, epithelial signalling, neuronal excitability and pain modulation. But under hypoxia, mechanical stress, epithelial injury, sympathetic firing, infection, mast-cell activation or mitochondrial strain, extracellular ATP may become excessive. High local extracellular ATP can activate P2X7, P2X4 and related P2Y signalling across mast cells, microglia, macrophages, neutrophils, epithelial cells, endothelial cells, nociceptors, astrocytes, fibroblasts and sympathetic varicosities. This biases the system toward calcium influx, potassium efflux, inflammasome activation, ROS production, cytokine release, degranulation, pain amplification and cell-danger signalling.
[see figure 19]
Normally, extracellular ATP is degraded through ADP and AMP toward adenosine and inosine, shifting the signal from danger toward resolution. In this model, that gating becomes unreliable. Intracellular purine synthesis may be constrained by low PRPP, impaired NAD⁺ synthesis, poor phosphate availability and redox stress, while extracellular ATP signalling remains high. This creates a pathological split: too little usable purine metabolism inside the cell, too much danger signalling outside the cell. Inosine and adenosine signalling, which should help terminate inflammatory signalling and support parasympathetic recovery, may become insufficient, mistimed or overwhelmed by persistent extracellular ATP relay.
[see figure 19]
The microbiome is then not a separate cause, but part of the same adaptive loop. Under sympathetic excess, hypoxia, altered motility, impaired bile flow, low glycogen reserve, immune suppression and redox stress, microbial communities may shift toward fermentation-dominant metabolism. Ethanol-producing and acetaldehyde-producing organisms may expand. Hydrogen sulphide, D-lactate, ammonia, endogenous opioid-like compounds and other microbial metabolites may increase. Some of these products may initially buffer stress or provide alternate signalling routes, but chronically they can worsen NAD⁺ depletion, aldehyde burden, hypoxia signalling, glutamate dysregulation, immune tolerance and mitochondrial inhibition.
[see figure 1]
Acetaldehyde is treated as a major amplifier in this model. It is not merely a toxic by-product. Chronic acetaldehyde exposure from dysbiosis, impaired alcohol metabolism or endogenous fermentation may burden ALDH1A1, ALDH1A2, ALDH2 and related aldehyde-detoxification pathways. As ALDH activity becomes overused or impaired, NAD⁺ redox worsens, and impaired NAD⁺ redox further reduces aldehyde clearance. This creates a direct feedback loop: acetaldehyde impairs NAD⁺ handling, and low NAD⁺ handling impairs acetaldehyde clearance.
[see figure 14]
Acetaldehyde also helps explain why the model predicts broad nutrient dysfunction rather than neat isolated deficiencies. It may lower zinc availability, impair magnesium-dependent reactions, form thiamine adducts and reduce functional TPP availability. It may interfere with riboflavin conversion toward FMN and FAD, disrupt intracellular B6 handling despite apparently elevated serum P5P, impair B5-to-CoA metabolism, disturb folate and B12 handling, lower SAMe, reduce retinoic acid signalling, impair CoQ10 synthesis and worsen BH4 oxidation. The result is a pseudo-deficiency state, where supplementation may raise blood levels without reliably restoring intracellular enzyme function.
[see figure 7]
This aldehyde-nutrient layer connects directly to histamine. Dysbiosis, epithelial barrier dysfunction, antigen exposure, extracellular ATP signalling and oxidative stress may all promote mast-cell degranulation. Mast cells then release histamine, while acetaldehyde, low zinc, oxidative stress and impaired aldehyde metabolism may reduce histamine clearance through DAO and ALDH-related mechanisms. Histamine therefore rises not only because mast cells are activated, but because clearance is impaired. Oestradiol and androgen status may further modulate this response, increasing sensitivity in some contexts.
[see figure 9]
Histamine then becomes a metabolic stress signal rather than just an allergy mediator. H1 and H2 signalling in the liver can promote glycogenolysis and contribute to liver glycogen depletion. Histamine can potentiate beta-adrenergic gain, increasing the probability of rebound hypoglycaemia, adrenergic surges, POTS-like physiology and post-exertional malaise. Perivascular histamine signalling may promote IL-6 release, STAT3 activation in myofibres and abnormal muscle glucose handling, including increased GLUT4 and glycogen-synthase signalling without effective glycogen restoration. In this state, exercise-induced histamine, which may be adaptive in a healthy system, becomes part of the post-exertional inflammatory and metabolic trap.
[see figure 9]
The redox chemistry reinforces the same loop. Acetaldehyde and inflammatory ROS may oxidise BH4, uncoupling eNOS and increasing superoxide production. Nitric oxide and superoxide may then form peroxynitrite, which can inhibit mitochondrial complex I and worsen NAD⁺/NADH redox. As NAD⁺ redox worsens, ALDH function worsens, acetaldehyde rises, histamine clearance falls and mitochondrial redox becomes more constrained. This links acetaldehyde, histamine intolerance, nitric oxide dysfunction, peroxynitrite stress and mitochondrial inhibition into one reinforcing circuit.
[see figure 1]
Once this threshold is crossed, sympathetic compensation becomes maladaptive. Initially, sympathetic activation is protective. It mobilises glucose, maintains blood pressure, increases alertness and attempts to preserve perfusion. But if glycogen storage, insulin signalling, NAD⁺ recovery, parasympathetic metabolism and glutamate clearance remain impaired, the same rescue response becomes a trap. Catecholamine signalling, cAMP-PKA-CREB activation, intracellular calcium stress, extracellular ATP release, histamine signalling and glutamate release can begin to amplify each other. The organism is driven to mobilise energy it cannot properly store, oxidise or recover from.
[see figure 10]
This also explains the apparent paradox of sympathetic overdrive alongside impaired intracellular signalling. The system may be adrenergically overactivated at the organism level, while specific downstream pathways such as glycogen synthesis, cAMP-PKA regulation, CREB signalling, NMNAT-dependent NAD⁺ synthesis and parasympathetic restoration remain impaired or mistimed. Endogenous opioid-like metabolites, acetaldehyde-derived signalling and chronic μ-opioid receptor activation may further distort adenylate cyclase and cAMP-PKA dynamics. The result is not simply “high sympathetic tone”, but noisy, dysregulated stress signalling that fails to produce stable metabolic recovery.
[see figure 1]
Connective tissue phenotypes, including hEDS-like or vEDS-like metabolism, are not peripheral in this model. Collagen synthesis and degradation depend on oxygen, iron, vitamin C, copper, silica, glycine, proline, lysine, redox balance, mitochondrial ATP and prolyl hydroxylase activity. Acidaemia, lactate accumulation, hypoxia signalling, oxidative stress, low mineral reserve, impaired NADPH repair and inflammatory cytokines may all disrupt collagen maintenance. This can contribute to vascular tone instability, gut barrier dysfunction, altered lymphatic flow, joint instability, mechanosensory amplification, pain and increased susceptibility to autonomic dysregulation. The connective tissue phenotype then feeds back into the model through mechanical stress, epithelial vulnerability, mast-cell activation and extracellular ATP release.
[see figure 12]
The immune system eventually becomes both overactive and ineffective. Early inflammatory signalling may be strong, but chronic redox failure, mineral misallocation, low ATP availability, cortisol dysregulation, impaired interferon responses, poor sleep and nutrient activation failure may reduce pathogen clearance. Latent viruses, bacterial reservoirs, fungal overgrowth or other microbial niches may persist. These reservoirs then periodically trigger inflammatory cascades, cytokine release, hepcidin activation, extracellular ATP signalling and mast-cell activation, further accelerating the original spiral.
[see figure 12]
This is why the “what came first?” question becomes less useful as the disease progresses. In early disease, it may matter whether the dominant initiating factor was infection, toxic metals, connective tissue fragility, neurodevelopmental stress load, pregnancy, nutrient insufficiency, microbiome shift, trauma, histamine excess or immune activation. In established disease, these factors become mutually reinforcing. Acidaemia worsens mineral wasting. Mineral wasting worsens ATP chemistry. Poor ATP chemistry worsens glutathione synthesis. Poor glutathione status worsens metal retention. Metal retention worsens mitochondrial redox. Redox failure worsens aldehyde clearance. Acetaldehyde worsens histamine clearance. Histamine worsens glycogen instability. Glycogen instability worsens sympathetic overdrive. Sympathetic overdrive increases extracellular ATP release. Extracellular ATP sustains cell-danger signalling. Cell-danger signalling sustains inflammation and hepcidin. The system becomes circular.
[see figure 10]
The persistence problem is therefore different from a simple vitamin deficiency, calorie deficit or macronutrient imbalance. Vitamins and macronutrients turn over relatively quickly. Elemental distribution does not. Electrolytes, minerals and toxic metals are stored, transported, sequestered and redistributed through inflammatory, renal, hepatic, mitochondrial, metallothionein, glutathione and membrane transport systems. Once inflammatory signalling and redox control are disturbed, it becomes progressively harder to move essential minerals into the compartments where they are needed and toxic metals out of the compartments where they are harmful.
[see figure 12]
In this model, chronic disease persistence is maintained by a locked state: low functional minerals, impaired ATP chemistry, poor NAD⁺ and NADPH redox, low glutathione capacity, metal excess and misallocation, acetaldehyde burden, histamine excess, extracellular ATP danger signalling, sympathetic overdrive, glutamate excess, connective tissue instability, microbiome fermentation, immune suppression and recurrent inflammatory triggering. The deeper the system moves into this cascade, the less effective isolated interventions become. Adding a vitamin, increasing calories, treating a pathogen, suppressing inflammation, supporting methylation or replacing a single mineral may help briefly, but may fail unless the broader transport and redox state is shifted.
[see figure 12]
The therapeutic implication is that recovery would require coordinated restoration of compartmental control, not merely replacement of missing inputs. The aim would be to reopen mineral transport, restore mineral status, remove toxic metals and re-establish functional daily nutrition, allowing the system to rebuild glutathione synthesis and recycling, restore NAD⁺ and NADPH balance, reduce aldehyde and peroxynitrite burden, improve glycogen storage, normalise histamine clearance, lower excessive extracellular ATP danger signalling, restore adenosine and inosine resolution pathways, reduce maladaptive sympathetic drive, rebuild parasympathetic metabolism, stabilise collagen repair and gradually reverse maladaptive microbiome fermentation and pathogen accumulation. The model’s central claim is that the system is not necessarily permanently broken, but trapped in a defensive metabolic configuration that becomes increasingly self-sustaining unless the mineral-redox-purinergic-histamine-autonomic-immune loop is interrupted.
Figures:
Figure 1 (click image to download PDF) or use the Metabolic Pathway Overlay tool to view this diagram with optional data overlay:
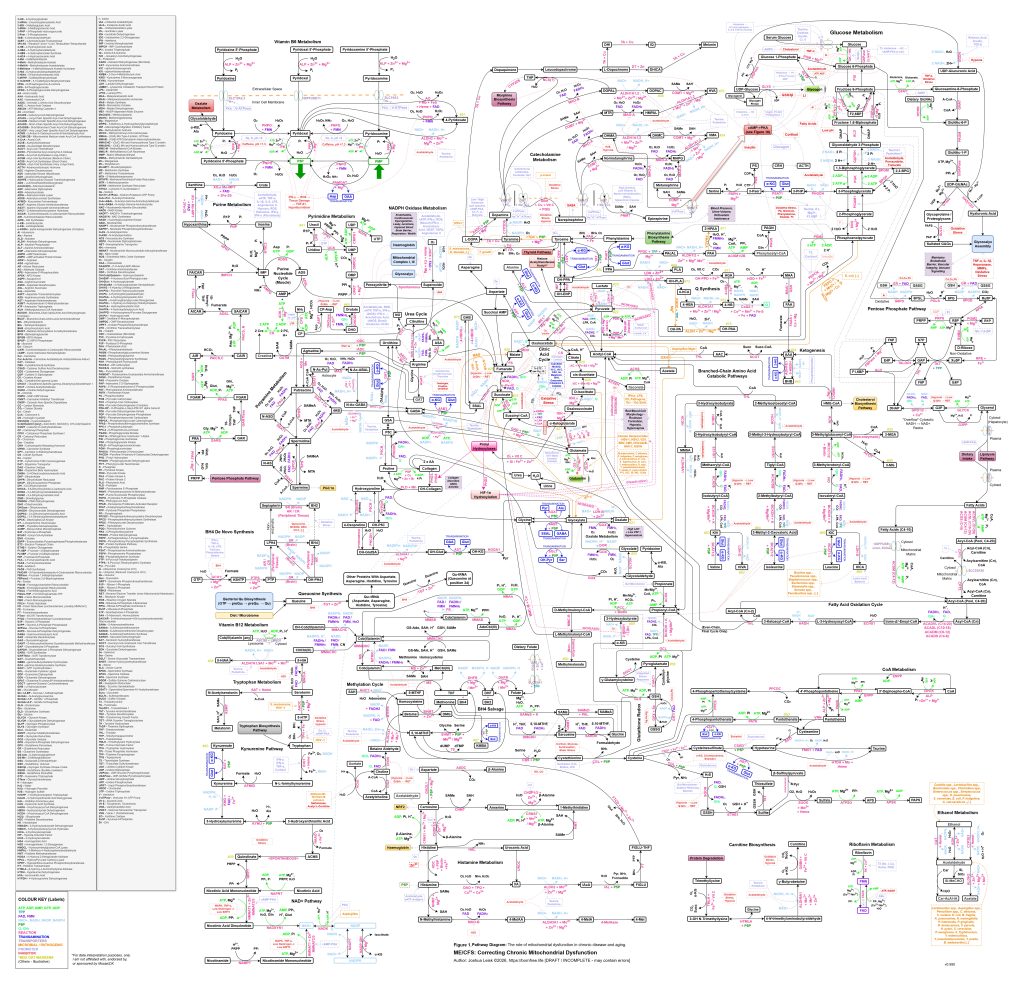
Figure 2 (legacy material - being retired/superseded by Figure 3 and will be replaced soon):
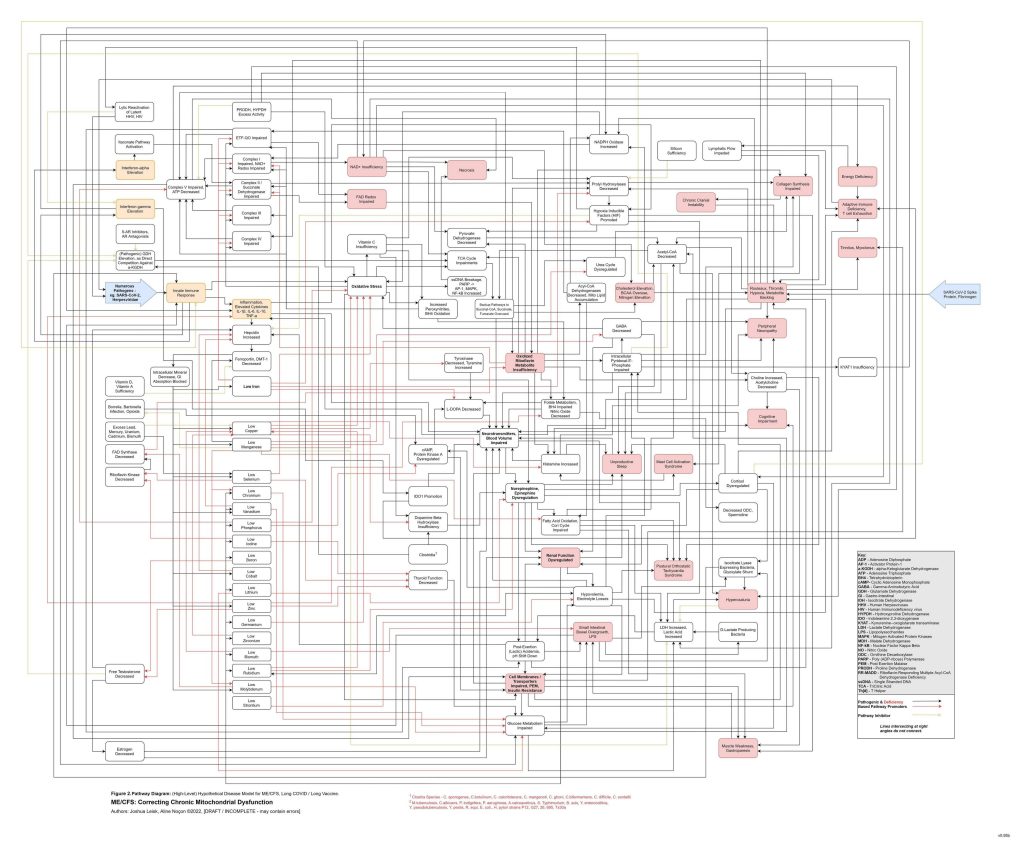
Figure 3: (Interactive explorer - here)
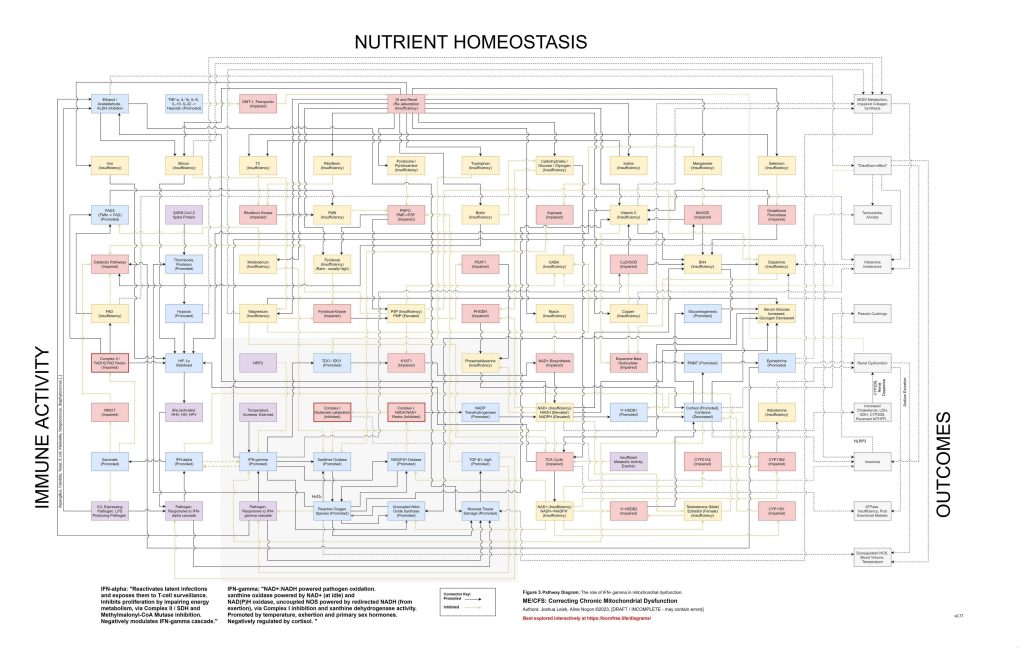
Figure 4 (click image to download PDF and interactive explorer - here)
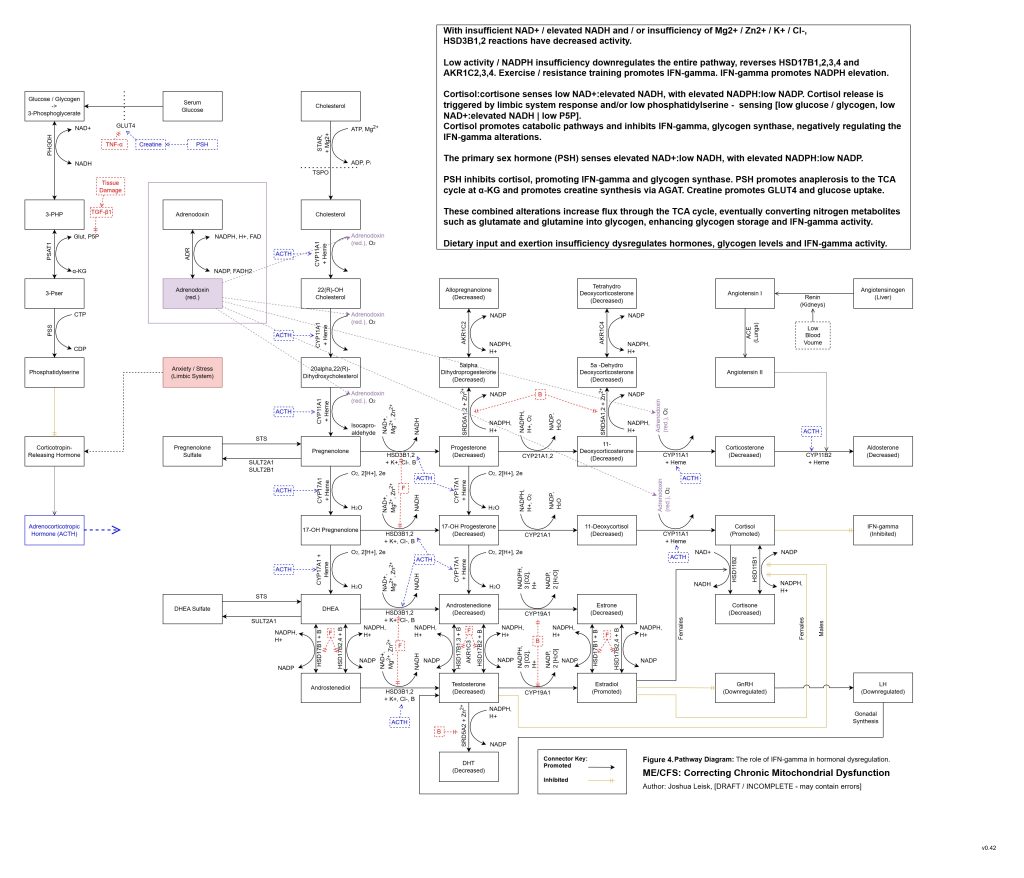
Figure 5 (Interactive explorer - here):
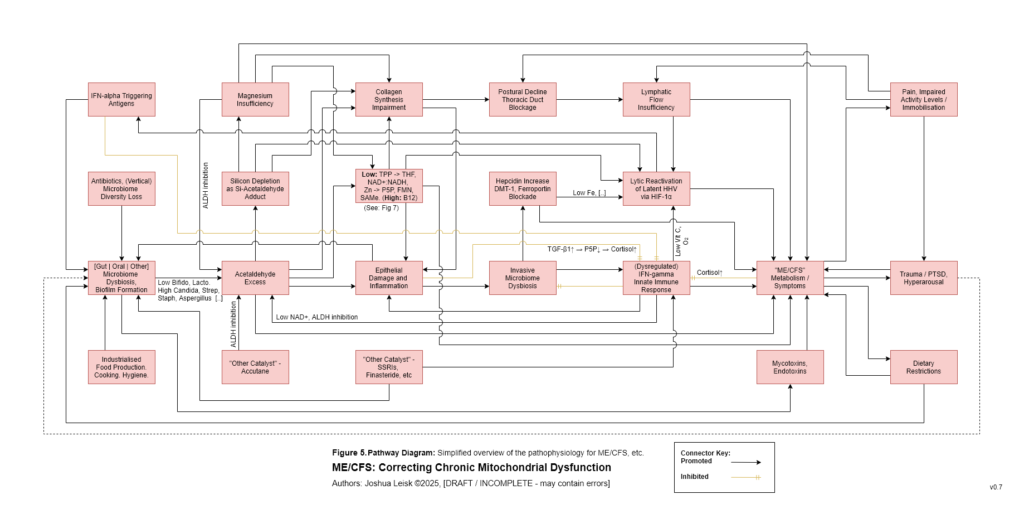
Figure 6 (click image to download PDF):
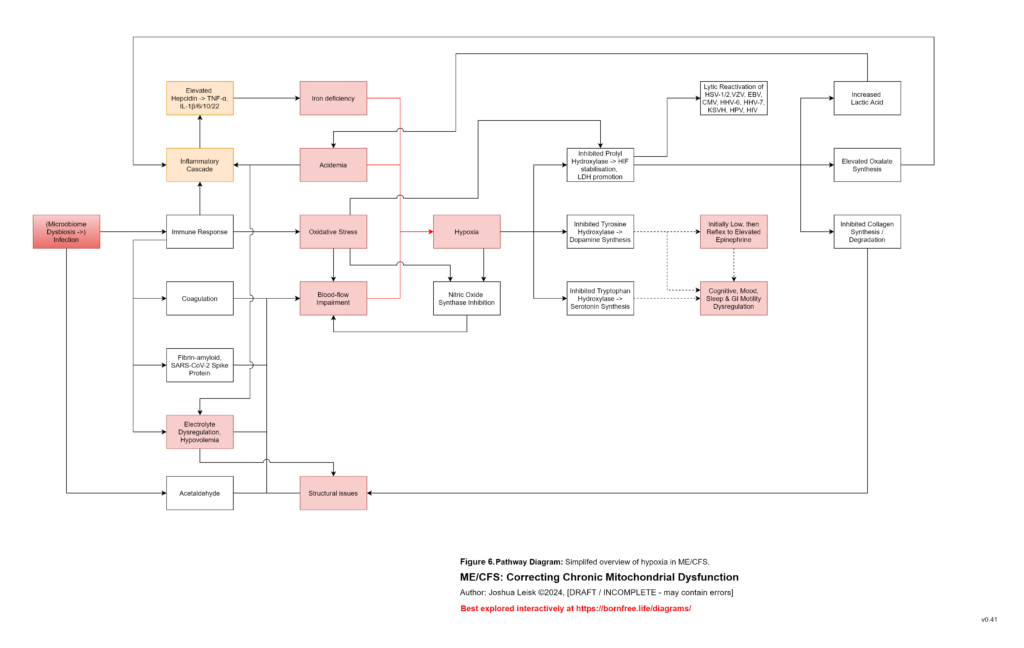
Figure 7 (click image to download PDF):
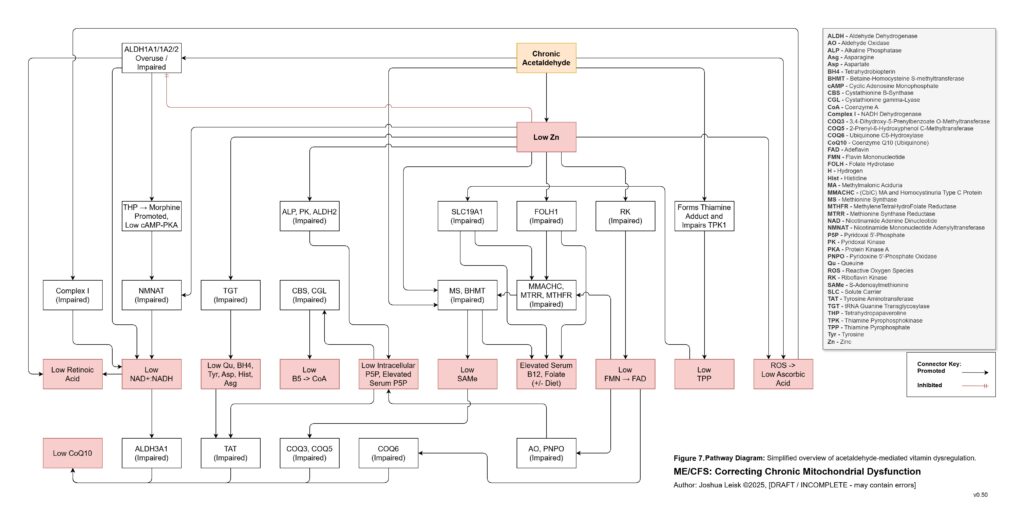
Figure 8 (click image to download PDF):
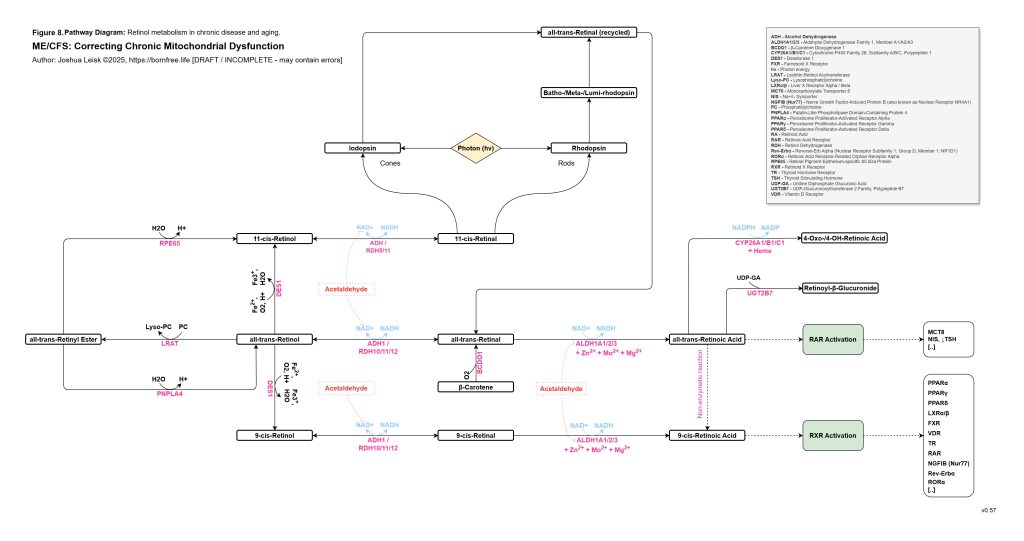
Figure 9 (click to download PDF):
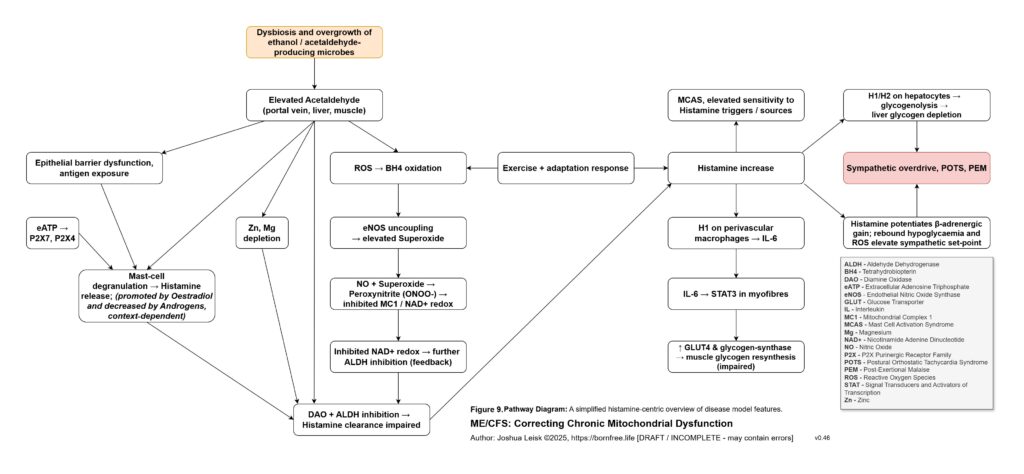
Figure 10 (click image to download PDF):
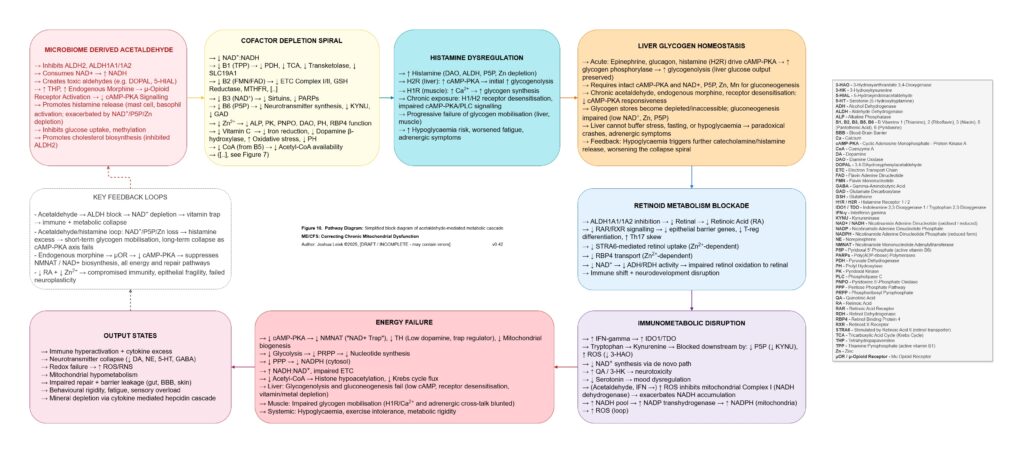
Figure 11 (click image to download PDF):
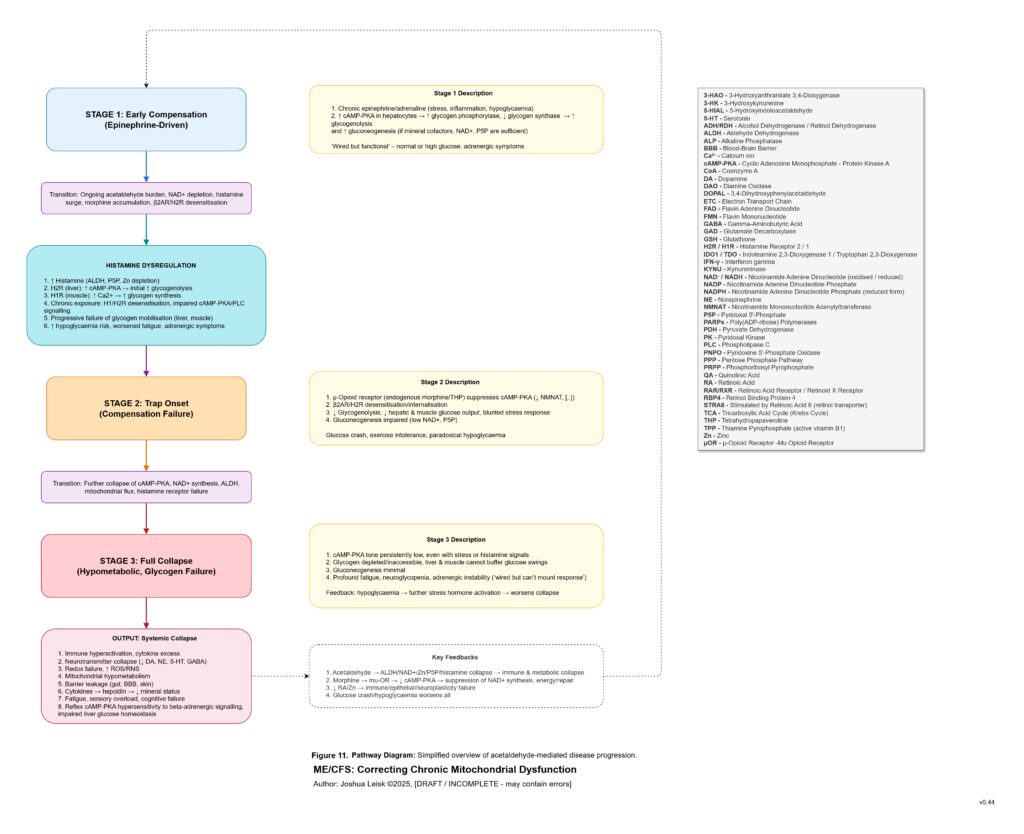
Figure 12 (click image to download PDF):
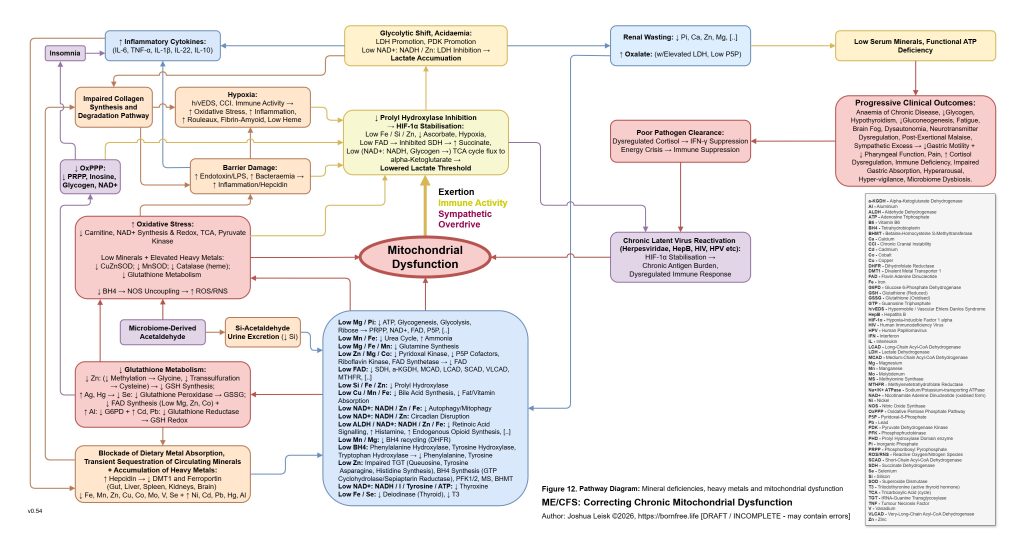
Figure 13 (click image to download PDF):
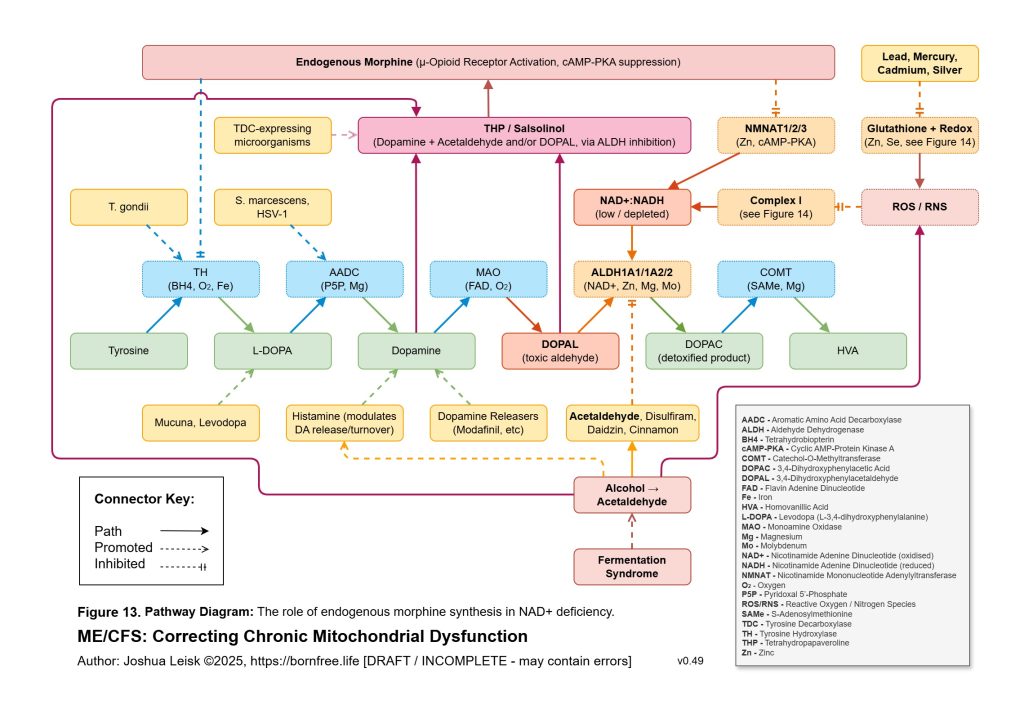
Figure 14 (click image to download PDF):
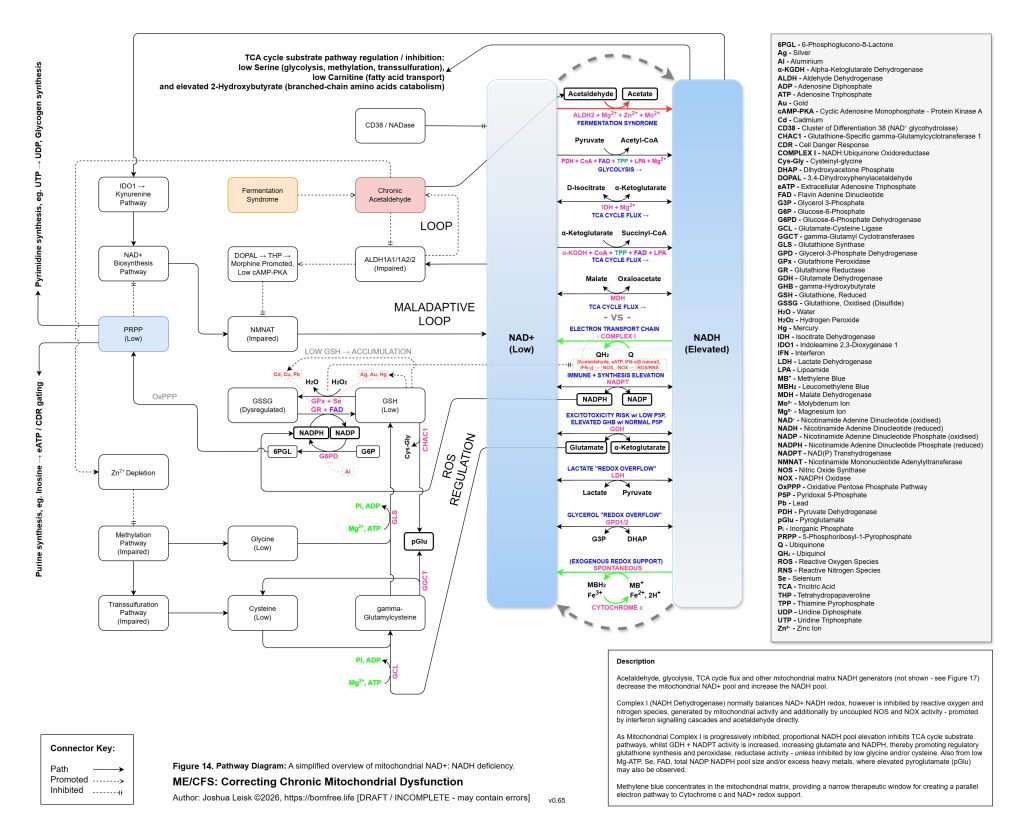
Figure 15 (click image to download PDF):
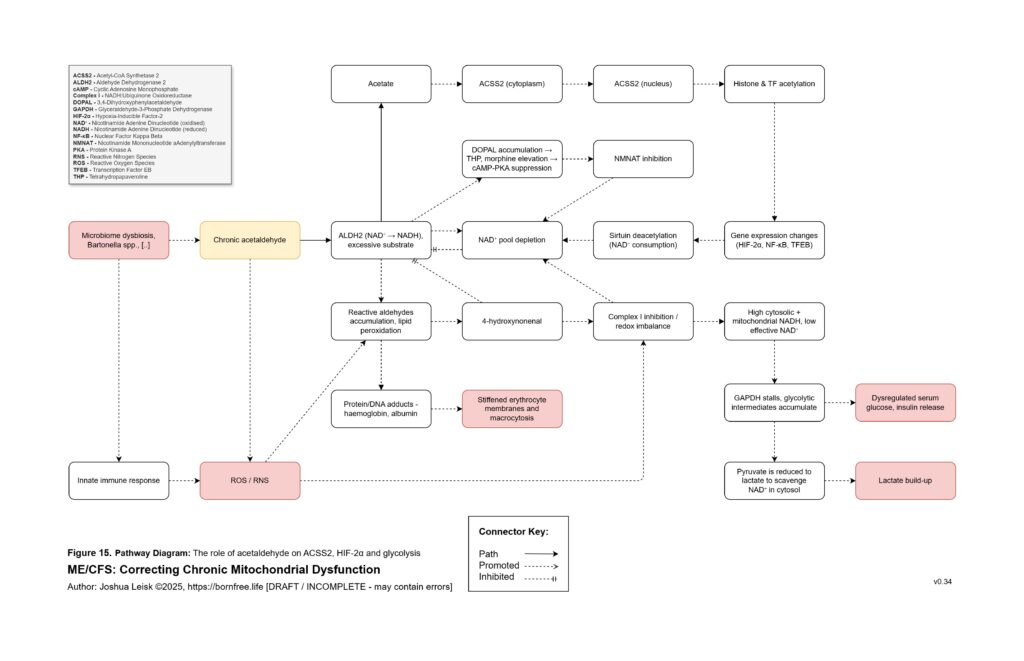
Figure 16 (click image to download PDF):
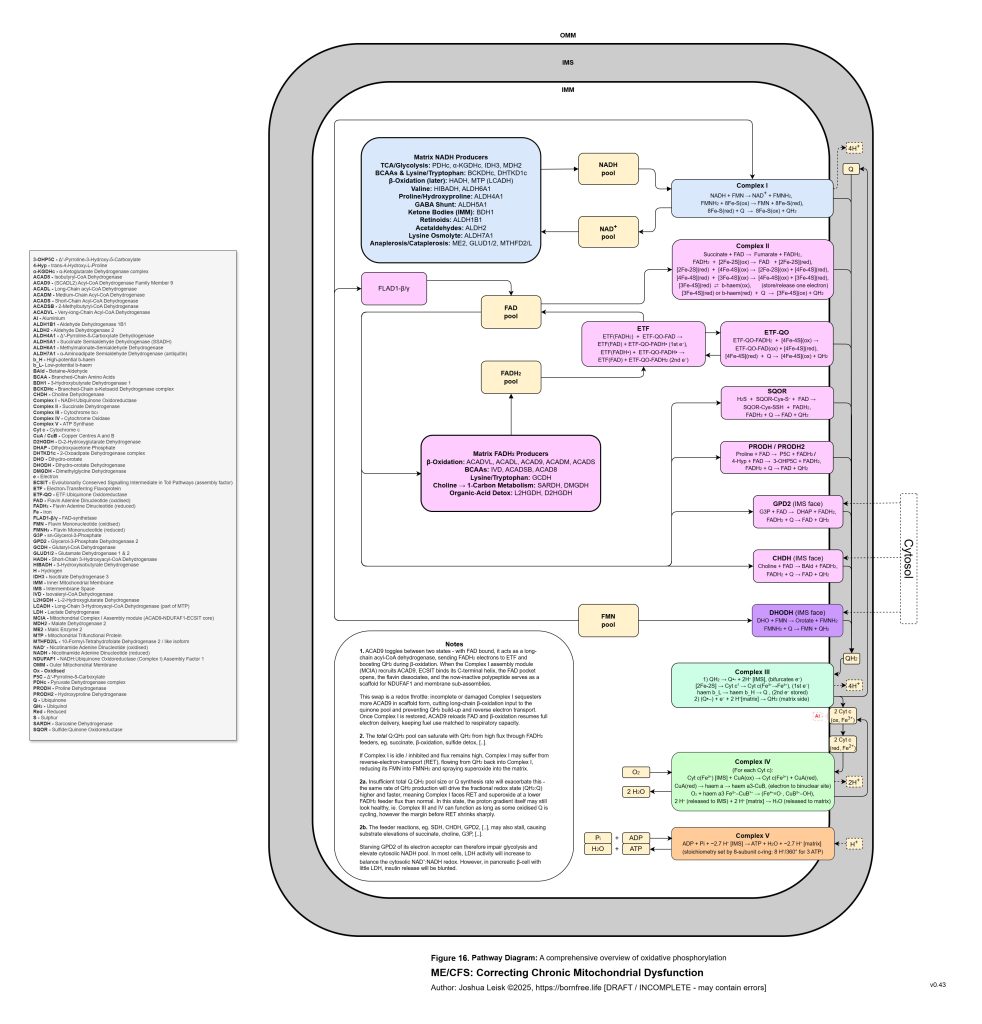
Figure 17 (click image to download PDF):
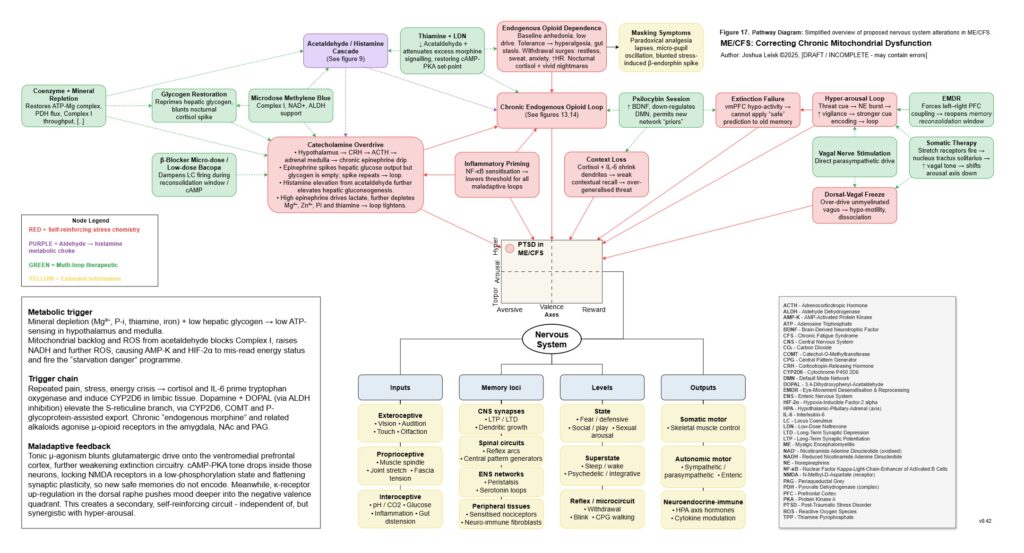
Figure 18 (click image to download PDF):
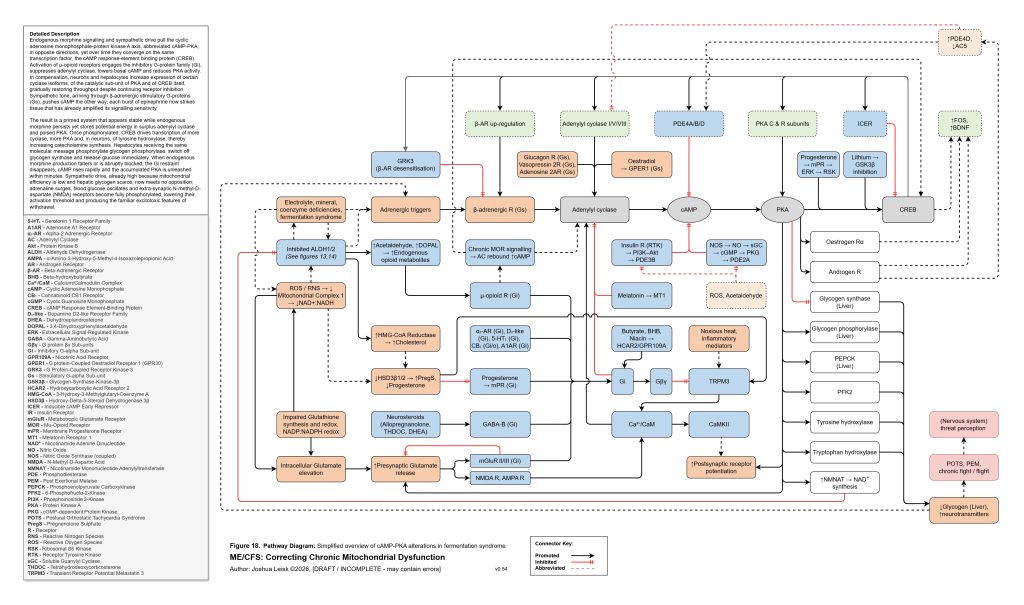
Figure 19 (click image to download PDF):